Health Tidings ResearchFDA Moves Toward Real-World Evaluation of Digital Devices With TEMPO Progra... FDA Moves Toward Real-World Evaluation of Digital Devices With TEMPO Progra... Read Post »
Clinical TrailsJnJ’s INLEXZO™ Achieves 74% One-Year Disease-Free Survival in BCG-U... JnJ’s INLEXZO™ Achieves 74% One-Year Disease-Free Survival in BCG-U... Read Post »
Clinical TrailsBayer Launches Phase III SUNFLOWER Trial: Mirena 52mg LNG-IUS for Nonatypic... Bayer Launches Phase III SUNFLOWER Trial: Mirena 52mg LNG-IUS for Nonatypic... Read Post »
Clinical TrailsPositive HOPE-3 Phase 3 Results Support Deramiocel as a Potential New Treat... Positive HOPE-3 Phase 3 Results Support Deramiocel as a Potential New Treat... Read Post »
Health Tidings ResearchAbbott Launches Two New Ensure® Max Protein Shakes: 42g Protein and 2-in-1... Abbott Launches Two New Ensure® Max Protein Shakes: 42g Protein and 2-in-1... Read Post »
New Drug ApprovalFDA Approves Breyanzi as First CAR T-Cell Therapy for Relapsed/Refractory M... FDA Approves Breyanzi as First CAR T-Cell Therapy for Relapsed/Refractory M... Read Post »
Clinical Trails New Drug ApprovalIonis’ Zilganersen Earns FDA Breakthrough Therapy Designation for Ultra R... Ionis’ Zilganersen Earns FDA Breakthrough Therapy Designation for Ultra R... Read Post »
Clinical TrailsLEQEMBI® (Lecanemab-irmb) CTAD 2025 Data Confirms Aβ Protofibril Reductio... LEQEMBI® (Lecanemab-irmb) CTAD 2025 Data Confirms Aβ Protofibril Reductio... Read Post »
New Drug ApprovalSun Pharma Launches Ilumya® in India: A Global Innovative Drug for Moderat... Sun Pharma Launches Ilumya® in India: A Global Innovative Drug for Moderat... Read Post »
New Drug ApprovalBaxdrostat NDA Accepted Under FDA Priority Review for Hard-to-Control High ... Baxdrostat NDA Accepted Under FDA Priority Review for Hard-to-Control High ... Read Post »
Drugs Safety Alert ResearchFDA Seizes 73,000 Illegal 7-OH Opioid Products worth $1M in Missouri: Major... FDA Seizes 73,000 Illegal 7-OH Opioid Products worth $1M in Missouri: Major... Read Post »
Drugs Safety Alert ResearchFDA Final Guidance on QTc Information in Human Prescription Drug and Biolog... FDA Final Guidance on QTc Information in Human Prescription Drug and Biolog... Read Post »
New Drug ApprovalAmneal Secure FDA Nod for Albuterol Inhaler and Cyclosporine Eye Drops in E... Amneal Secure FDA Nod for Albuterol Inhaler and Cyclosporine Eye Drops in E... Read Post »
Clinical TrailsEisai Unveils Breakthrough Etalanetug Data at CTAD 2025: Significant Reduct... Eisai Unveils Breakthrough Etalanetug Data at CTAD 2025: Significant Reduct... Read Post »
New Drug ApprovalUS FDA Accepts Wockhardt’s NDA for Zaynich, Marking a Historic First for ... US FDA Accepts Wockhardt’s NDA for Zaynich, Marking a Historic First for ... Read Post »
Clinical TrailsMerck Presents Promising First-in-Human Data for Alzheimer’s Candidates M... Merck Presents Promising First-in-Human Data for Alzheimer’s Candidates M... Read Post »
Clinical TrailsAbbVie’s Phase 3 ECLIPSE Study Shows Atogepant (AQUIPTA®) Superior t... AbbVie’s Phase 3 ECLIPSE Study Shows Atogepant (AQUIPTA®) Superior t... Read Post »
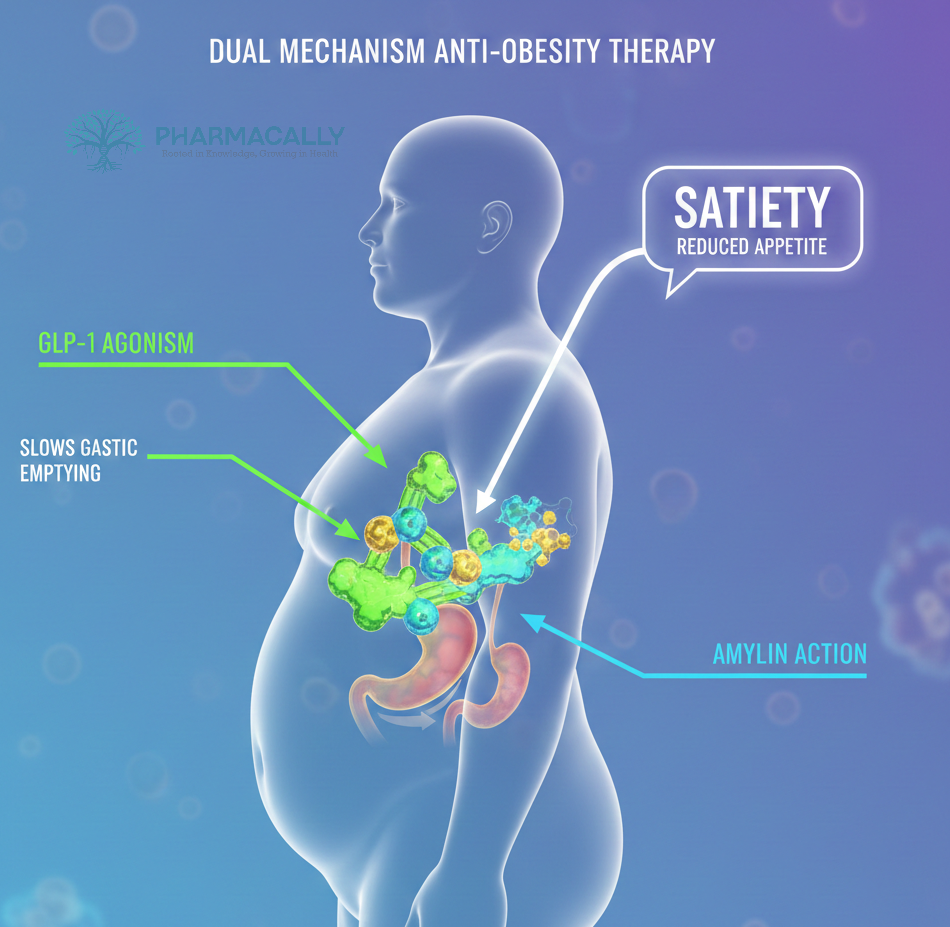
Health Tidings ResearchEli Lilly Lowers Single-Dose Zepbound Vial Prices Eli Lilly Lowers Single-Dose Zepbound Vial Prices Read Post »
Drugs Safety Alert ResearchAustralia’s TGA Updates Safety Warnings for GLP-1 Receptor Agonists a... Australia’s TGA Updates Safety Warnings for GLP-1 Receptor Agonists a... Read Post »
ResearchWHO Measles Report 2025: Vaccination Triumphs save Millions but Outbreaks W... WHO Measles Report 2025: Vaccination Triumphs save Millions but Outbreaks W... Read Post »
Clinical TrailsOral Mosnodenvir Pill’s Rocky Road: 60% Dengue Success in Phase 2a Tr... Oral Mosnodenvir Pill’s Rocky Road: 60% Dengue Success in Phase 2a Tr... Read Post »
New Drug ApprovalChina’s First Domestic IL-23p19 Antibody PECONDLE Gains NMPA Approval... China’s First Domestic IL-23p19 Antibody PECONDLE Gains NMPA Approval... Read Post »
Clinical Trails Health TidingsAlexion Advances ALXN2420 to Phase 2: PeptiDream Unlocks Acromegaly Milesto... Alexion Advances ALXN2420 to Phase 2: PeptiDream Unlocks Acromegaly Milesto... Read Post »
New Drug ApprovalTreating ADHD Across Lifespan: Otsuka’s Centanafadine NDA Submission Insi... Treating ADHD Across Lifespan: Otsuka’s Centanafadine NDA Submission Insi... Read Post »
New Drug ApprovalEisai Completes Fast Track sBLA to FDA and Submits NDA to Japan PMDA for Su... Eisai Completes Fast Track sBLA to FDA and Submits NDA to Japan PMDA for Su... Read Post »
New Drug ApprovalFDA Accelerates Approval of Otsuka’s VOYXACT® (sibeprenlimab-szsi) for I... FDA Accelerates Approval of Otsuka’s VOYXACT® (sibeprenlimab-szsi) for I... Read Post »
Clinical Trails New Drug ApprovalNovo Nordisk Files FDA sNDA for Wegovy 7.2 mg Higher Dose After 20.7% Weigh... Novo Nordisk Files FDA sNDA for Wegovy 7.2 mg Higher Dose After 20.7% Weigh... Read Post »
New Drug ApprovalFDA Approves Imfinzi as First and Only Perioperative Immunotherapy for Earl... FDA Approves Imfinzi as First and Only Perioperative Immunotherapy for Earl... Read Post »
New Drug ApprovalSanofi and Regeneron’s Dupixent Wins EU Approval as First Targeted Therap... Sanofi and Regeneron’s Dupixent Wins EU Approval as First Targeted Therap... Read Post »
Drugs Safety Alert ResearchAbbott Initiates Medical Device Correction for Freestyle Libre 3 Sensors in... Abbott Initiates Medical Device Correction for Freestyle Libre 3 Sensors in... Read Post »
Clinical TrailsSarepta Receives FDA Approval to Begin ENDEAVOR Cohort 8 Investigating Enha... Sarepta Receives FDA Approval to Begin ENDEAVOR Cohort 8 Investigating Enha... Read Post »
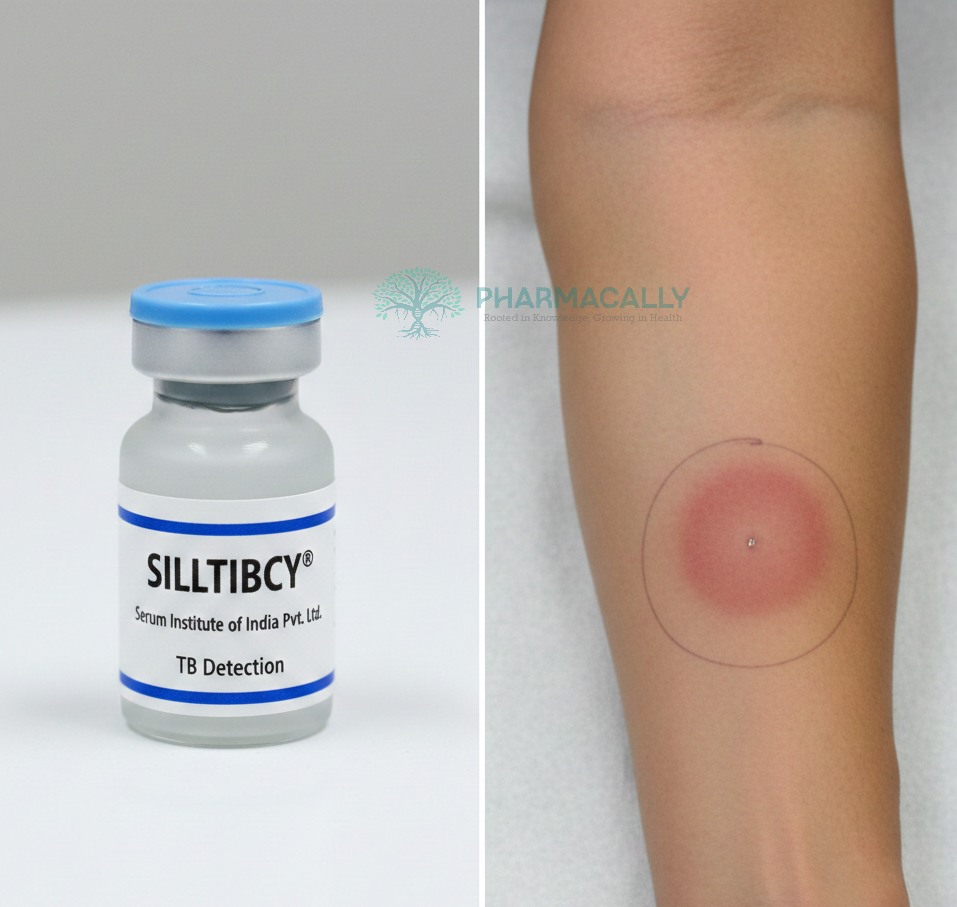
New Drug ApprovalSerum Institute of India’s SIILTIBCY® Receives MHRA Authorization in... Serum Institute of India’s SIILTIBCY® Receives MHRA Authorization in... Read Post »
Clinical TrailsNovo Nordisk’s Amycretin Phase 2 Trial Shows Up to 14.5% Weight Loss and ... Novo Nordisk’s Amycretin Phase 2 Trial Shows Up to 14.5% Weight Loss and ... Read Post »
New Drug ApprovalFDA Approves Itvisma (Onasemnogene Abeparvovec-brve), the First Intrathecal... FDA Approves Itvisma (Onasemnogene Abeparvovec-brve), the First Intrathecal... Read Post »
Clinical TrailsJohnson & Johnson’s Posdinemab Fails to Meet Primary Endpoint in Auτ... Johnson & Johnson’s Posdinemab Fails to Meet Primary Endpoint in Auτ... Read Post »
Clinical TrailsKyowa Kirin Launches Phase 3 AOBA Study of KK8398 (Infigratinib) for Achond... Kyowa Kirin Launches Phase 3 AOBA Study of KK8398 (Infigratinib) for Achond... Read Post »
Clinical TrailsSarepta’s SRP-1003 siRNA Program Hits Key Enrollment Milestone, Triggers ... Sarepta’s SRP-1003 siRNA Program Hits Key Enrollment Milestone, Triggers ... Read Post »
Clinical TrailsNovo Nordisk’s Evoke Phase 3 Trial Fails to Show Semaglutide Benefit in A... Novo Nordisk’s Evoke Phase 3 Trial Fails to Show Semaglutide Benefit in A... Read Post »
Clinical TrailsNew Hope for Stroke Survivors: Bayer’s Asundexian Shows Promise in Reduci... New Hope for Stroke Survivors: Bayer’s Asundexian Shows Promise in Reduci... Read Post »
Drugs Safety Alert ResearchThe Truth about “Ozempic Penis”: What Men Should Know About Semaglutide... The Truth about “Ozempic Penis”: What Men Should Know About Semaglutide... Read Post »
Health Tidings ResearchEli Lilly Hits $1 Trillion Market Cap: How Mounjaro and Zepbound Revolution... Eli Lilly Hits $1 Trillion Market Cap: How Mounjaro and Zepbound Revolution... Read Post »
New Drug ApprovalKoselugo Approved by FDA for Adults with Neurofibromatosis Type 1: A New Tr... Koselugo Approved by FDA for Adults with Neurofibromatosis Type 1: A New Tr... Read Post »
New Drug ApprovalFDA Approves KEYTRUDA® (pembrolizumab) and KEYTRUDA QLEX™ with Padcev® ... FDA Approves KEYTRUDA® (pembrolizumab) and KEYTRUDA QLEX™ with Padcev® ... Read Post »
Drugs Safety Alert ResearchFDA Investigates Fatal Case of Neutralizing Antibodies to ADAMTS13 after Ad... FDA Investigates Fatal Case of Neutralizing Antibodies to ADAMTS13 after Ad... Read Post »
New Drug ApprovalFDA Grants Full Approval to AbbVie’s EPKINLY® (epcoritamab-bysp) Plu... FDA Grants Full Approval to AbbVie’s EPKINLY® (epcoritamab-bysp) Plu... Read Post »
Clinical TrailsPfizer’s Next-Gen mRNA Flu Shot: A New Standard in Vaccine Innovation Pfizer’s Next-Gen mRNA Flu Shot: A New Standard in Vaccine Innovation Read Post »
Clinical TrailsAgios’ Mitapivat Delivers Positive Hemoglobin Gains but Mixed Pain‑Cr... Agios’ Mitapivat Delivers Positive Hemoglobin Gains but Mixed Pain‑Cr... Read Post »
Clinical TrailsMerck Announces Positive Topline Results from Pivotal Phase 3 Trial of Dora... Merck Announces Positive Topline Results from Pivotal Phase 3 Trial of Dora... Read Post »
New Drug ApprovalFDA Clears Regeneron and Bayer’s EYLEA HD® for Macular Edema after R... FDA Clears Regeneron and Bayer’s EYLEA HD® for Macular Edema after R... Read Post »
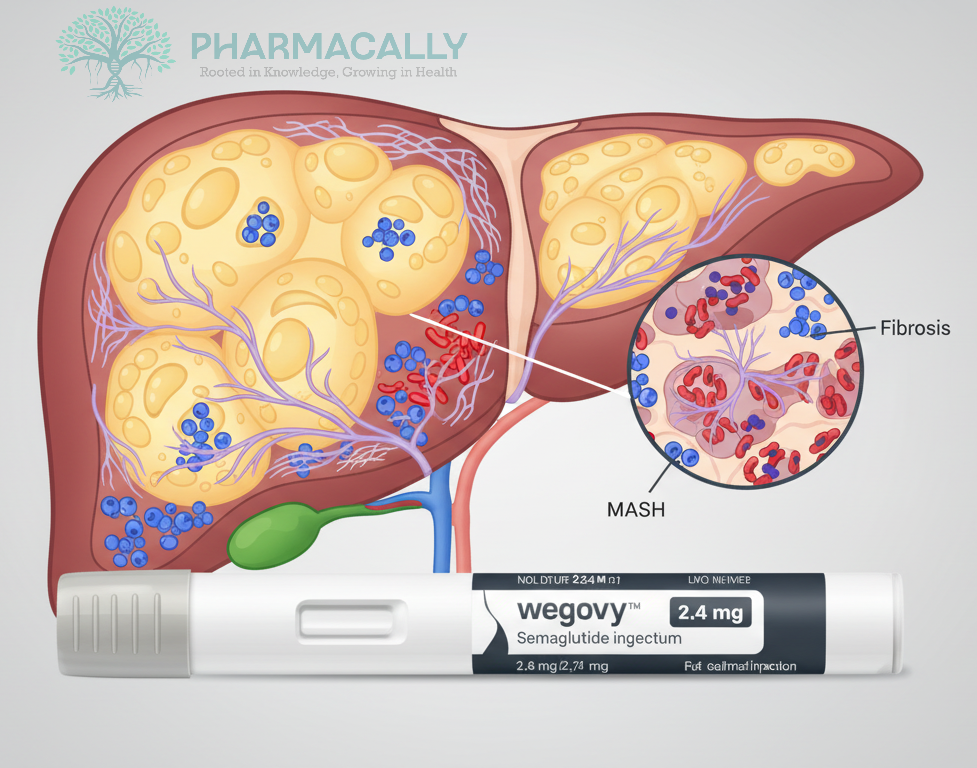
Clinical TrailsNovo Nordisk’s Wegovy® (Semaglutide 2.4 mg) Delivers Liver Health Benefi... Novo Nordisk’s Wegovy® (Semaglutide 2.4 mg) Delivers Liver Health Benefi... Read Post »