New Drug ApprovalGSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix GSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix Read Post »
New Drug ApprovalFDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment FDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment Read Post »
New Drug ApprovalFDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... FDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... Read Post »
New Drug ApprovalFDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... FDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... Read Post »
Clinical TrailsJohnson & Johnson’s RYBREVANT® Shows Promising Long-Term Results in ... Johnson & Johnson’s RYBREVANT® Shows Promising Long-Term Results in ... Read Post »
New Drug ApprovalSanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Sanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Read Post »
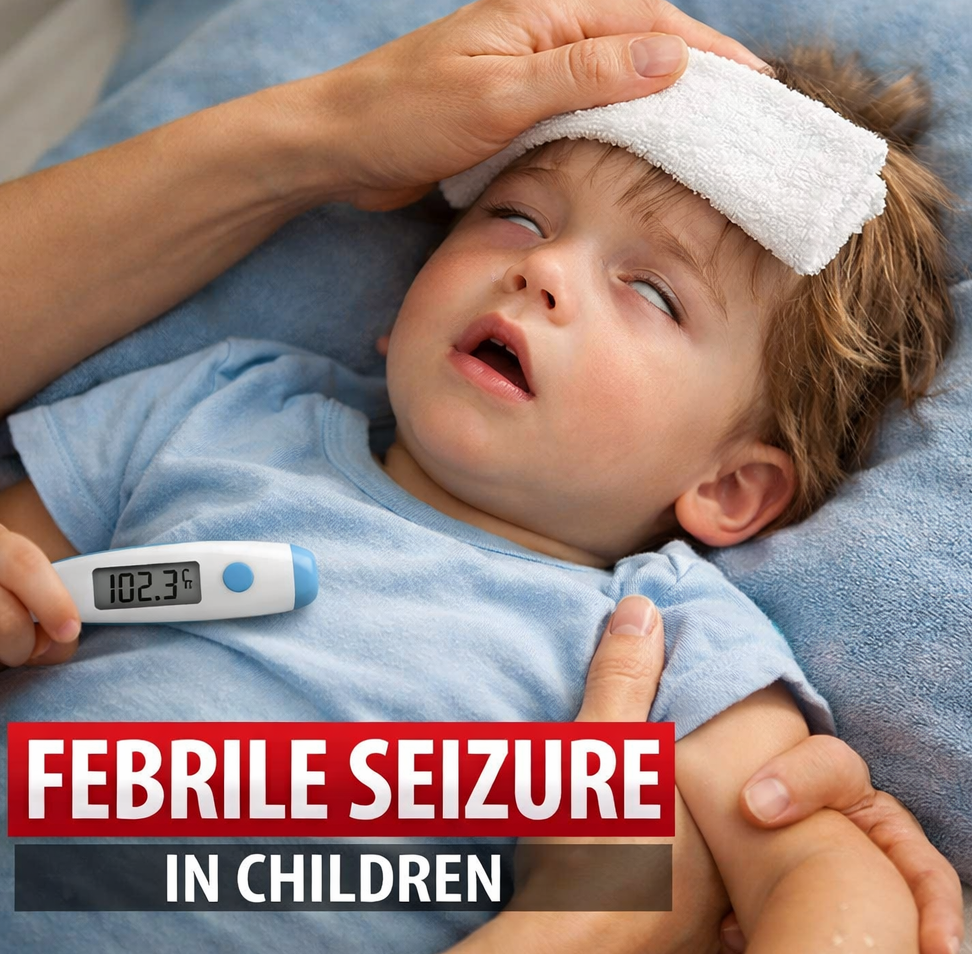
Drugs Safety AlertFDA CBER Flags Rare Febrile Seizure Risk Linked to Influenza Vaccines FDA CBER Flags Rare Febrile Seizure Risk Linked to Influenza Vaccines Read Post »
New Drug ApprovalBayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Bayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Read Post »
New Drug ApprovalChina Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... China Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... Read Post »
New Drug ApprovalMilestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Milestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Read Post »
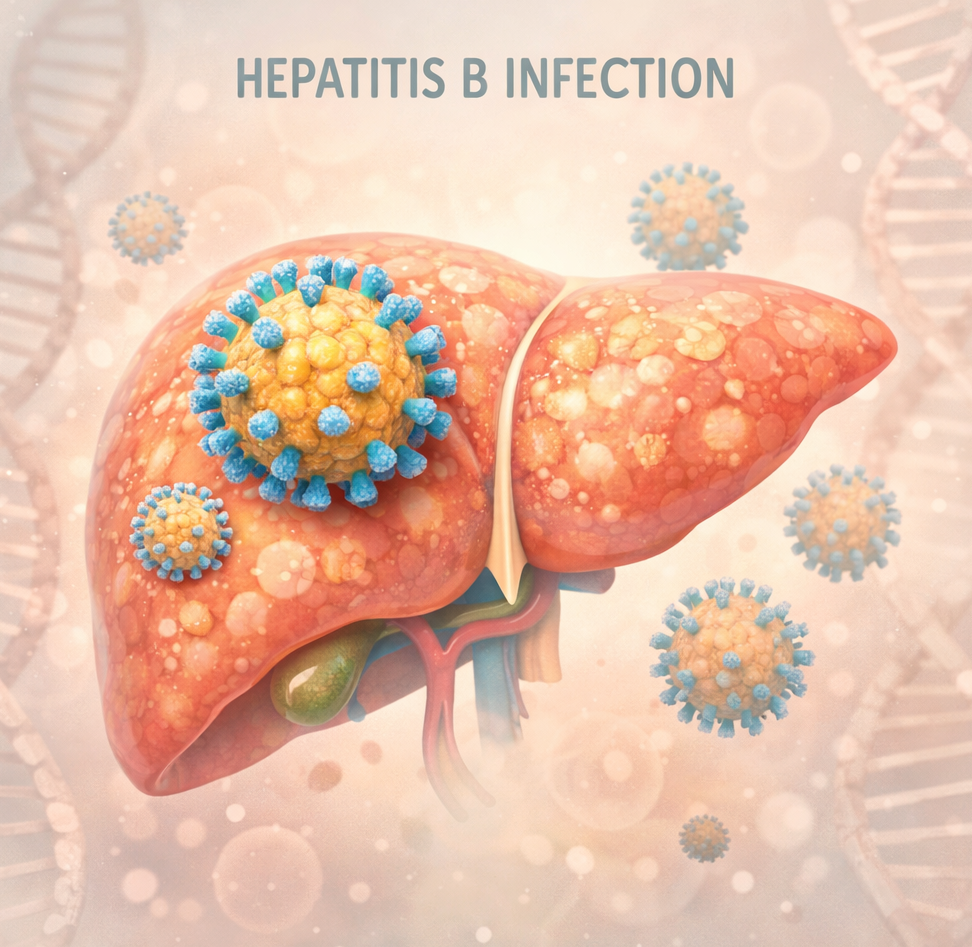
Clinical TrailsBepirovirsen Shows Phase III Success in Chronic Hepatitis B, GSK Confirms Bepirovirsen Shows Phase III Success in Chronic Hepatitis B, GSK Confirms Read Post »
Clinical TrailsLilly’s Taltz Plus Zepbound Delivers Superior Outcomes in Phase 3b Psoria... Lilly’s Taltz Plus Zepbound Delivers Superior Outcomes in Phase 3b Psoria... Read Post »
New Drug ApprovalFDA expands Cablivi approval to pediatric patients aged 12 and older with a... FDA expands Cablivi approval to pediatric patients aged 12 and older with a... Read Post »
New Drug ApprovalTakeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Takeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Read Post »
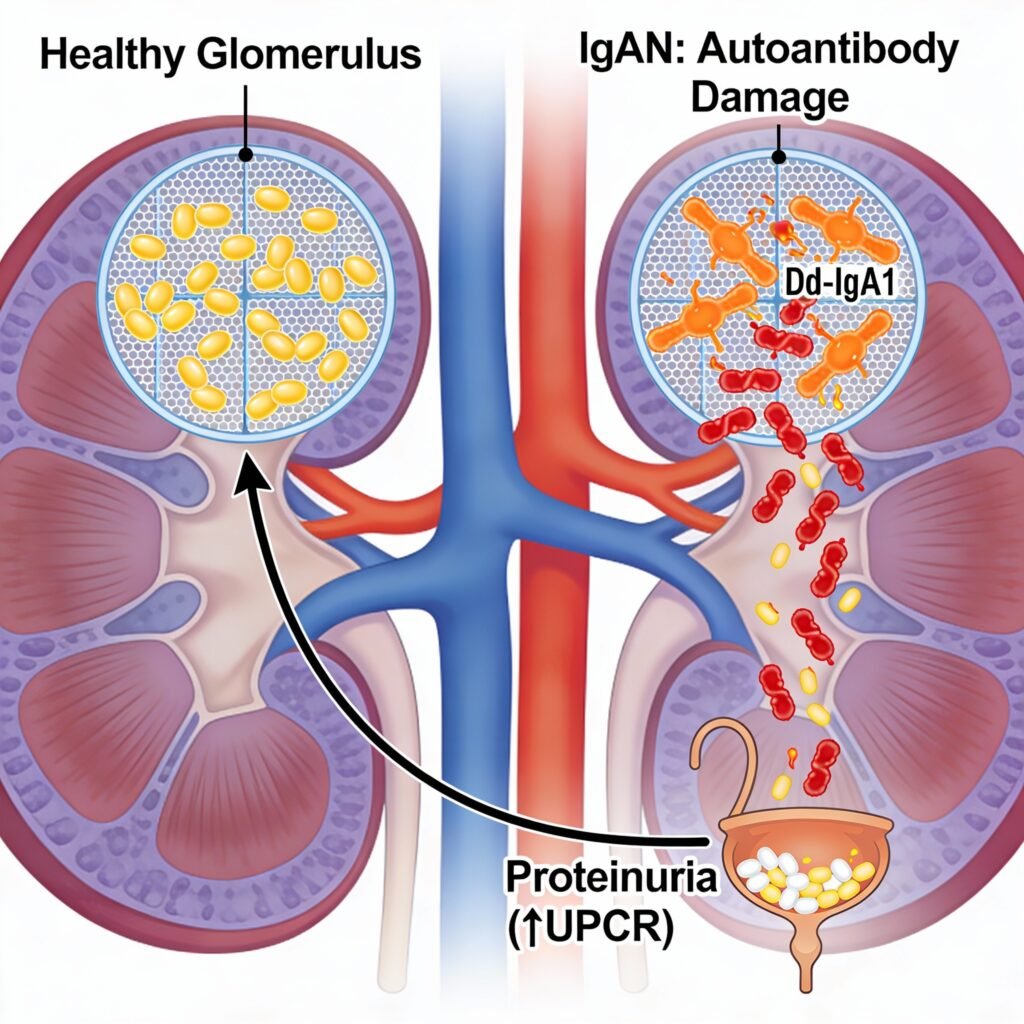
New Drug ApprovalFDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... FDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... Read Post »
Clinical TrailsJohnson & Johnson Reports Positive Phase 2b JASMINE Trial Results for N... Johnson & Johnson Reports Positive Phase 2b JASMINE Trial Results for N... Read Post »
New Drug ApprovalJapan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Japan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Read Post »
New Drug ApprovalZai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Zai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Read Post »
New Drug ApprovalChina Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s China Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s Read Post »
Clinical TrailsFull Phase 3 TULIP-SC Results Support Self-Administered Saphnelo for Lupus Full Phase 3 TULIP-SC Results Support Self-Administered Saphnelo for Lupus Read Post »
New Drug ApprovalHealth Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Health Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Read Post »
Health Tidings ResearchAbbott Launches Libre Assist in Libre App to Improve In-the-Moment Food Dec... Abbott Launches Libre Assist in Libre App to Improve In-the-Moment Food Dec... Read Post »
New Drug ApprovalModerna Files With FDA, EMA, Health Canada and TGA for New mRNA Flu Shot Moderna Files With FDA, EMA, Health Canada and TGA for New mRNA Flu Shot Read Post »
New Drug ApprovalAltimmune’s Pemvidutide Granted FDA Breakthrough Therapy Designation for ... Altimmune’s Pemvidutide Granted FDA Breakthrough Therapy Designation for ... Read Post »
New Drug ApprovalGSK’s Nucala (Mepolizumab) Gains China NMPA Approval for Eosinophilic... GSK’s Nucala (Mepolizumab) Gains China NMPA Approval for Eosinophilic... Read Post »
New Drug ApprovalSanofi’s Tzield (Teplizumab) Gains FDA Priority Review for 1-Year-Old... Sanofi’s Tzield (Teplizumab) Gains FDA Priority Review for 1-Year-Old... Read Post »
New Drug ApprovalMHRA Approves Kostaive (Zapomeran) sa-mRNA COVID Booster for UK Adults-Key ... MHRA Approves Kostaive (Zapomeran) sa-mRNA COVID Booster for UK Adults-Key ... Read Post »
New Drug ApprovalUltragenyx Completes Rolling BLA for First-in-Class DTX401 Gene Therapy in ... Ultragenyx Completes Rolling BLA for First-in-Class DTX401 Gene Therapy in ... Read Post »
ResearchPolyrizon Submits Pre-Request for Designation to FDA for PL-16 Viral Blocke... Polyrizon Submits Pre-Request for Designation to FDA for PL-16 Viral Blocke... Read Post »
Drugs Safety Alert ResearchNimesulide’s Rise and Reckoning in India: Ending with a Nationwide Ban on... Nimesulide’s Rise and Reckoning in India: Ending with a Nationwide Ban on... Read Post »
Drugs Safety AlertFDA Adds New Safety Warning to ALTUVIIIO Label FDA Adds New Safety Warning to ALTUVIIIO Label Read Post »
New Drug ApprovalFDA Accepts Axsome’s AXS-05 Supplemental NDA and Grants Priority Review f... FDA Accepts Axsome’s AXS-05 Supplemental NDA and Grants Priority Review f... Read Post »
Health Tidings ResearchFDA Issues Third CRL for Outlook Therapeutics’ ONS-5010/LYTENAVA in W... FDA Issues Third CRL for Outlook Therapeutics’ ONS-5010/LYTENAVA in W... Read Post »
ResearchSemaglutide and Eye Health: A Look at NAION Risk Semaglutide and Eye Health: A Look at NAION Risk Read Post »
Health Tidings ResearchCorcept Therapeutics Receives FDA Complete Response Letter for Relacorilant... Corcept Therapeutics Receives FDA Complete Response Letter for Relacorilant... Read Post »
New Drug ApprovalFDA Clears NEREUS, First New Motion Sickness Drug in Decades FDA Clears NEREUS, First New Motion Sickness Drug in Decades Read Post »
Clinical TrailsDenali’s Tividenofusp Alfa (DNL310) Phase 1/2 Data in Hunter Syndrome wit... Denali’s Tividenofusp Alfa (DNL310) Phase 1/2 Data in Hunter Syndrome wit... Read Post »
New Drug ApprovalUnicycive Resubmits NDA for Oxylanthanum Carbonate for Hyperphosphatemia Ma... Unicycive Resubmits NDA for Oxylanthanum Carbonate for Hyperphosphatemia Ma... Read Post »
New Drug ApprovalFDA Grants Breakthrough Therapy to Ulixacaltamide (PRAX-944) for Essential ... FDA Grants Breakthrough Therapy to Ulixacaltamide (PRAX-944) for Essential ... Read Post »
Health Tidings ResearchGenmab Halts Clinical Development of Acasunlimab After Portfolio Review, Sh... Genmab Halts Clinical Development of Acasunlimab After Portfolio Review, Sh... Read Post »
Drugs Safety Alert ResearchFDA Says PFAS Safety in Cosmetics Remains Uncertain Due to Data Gaps FDA Says PFAS Safety in Cosmetics Remains Uncertain Due to Data Gaps Read Post »
New Drug ApprovalFirst-in-Class TRPV1 Inhibitor Avarept Approved in Japan for Dry Eye Diseas... First-in-Class TRPV1 Inhibitor Avarept Approved in Japan for Dry Eye Diseas... Read Post »
New Drug Approval ResearchGrifols’ FESILTY (Fibrinogen): Evidence, Safety, and Clinical Use Explain... Grifols’ FESILTY (Fibrinogen): Evidence, Safety, and Clinical Use Explain... Read Post »
Clinical TrailsGalapagos GLPG3667 Meets Primary Endpoint in Dermatomyositis Phase 3-Enabli... Galapagos GLPG3667 Meets Primary Endpoint in Dermatomyositis Phase 3-Enabli... Read Post »
Clinical TrailsTirzepatide Matches Dulaglutide on Major Cardiovascular Outcomes in SURPASS... Tirzepatide Matches Dulaglutide on Major Cardiovascular Outcomes in SURPASS... Read Post »
Clinical TrailsJohnson and Johnson’s JNJ-5939 Falls Short in DUPLEX-AD Phase 2b Trial fo... Johnson and Johnson’s JNJ-5939 Falls Short in DUPLEX-AD Phase 2b Trial fo... Read Post »
New Drug ApprovalENHERTU Approved in China for HER2-Low and HER2-Ultralow Metastatic Breast ... ENHERTU Approved in China for HER2-Low and HER2-Ultralow Metastatic Breast ... Read Post »
New Drug ApprovalFDA Approves YARTEMLEA: First TA-TMA Therapy Post-Stem Cell Transplant FDA Approves YARTEMLEA: First TA-TMA Therapy Post-Stem Cell Transplant Read Post »
Clinical TrailsBiogen Reports Final VALOR Results Showing Earlier QALSODY Treatment May Sl... Biogen Reports Final VALOR Results Showing Earlier QALSODY Treatment May Sl... Read Post »
New Drug ApprovalIncyte’s Zynyz Receives First Approval in Japan for Advanced SCAC Incyte’s Zynyz Receives First Approval in Japan for Advanced SCAC Read Post »