New Drug ApprovalFDA Approves Tenpoint’s YUVEZZI Eye Drop for Presbyopia Treatment FDA Approves Tenpoint’s YUVEZZI Eye Drop for Presbyopia Treatment Read Post »
Clinical TrailsUltragenyx Resubmits BLA for UX111 Gene Therapy in Sanfilippo Syndrome Type... Ultragenyx Resubmits BLA for UX111 Gene Therapy in Sanfilippo Syndrome Type... Read Post »
Clinical TrailsTanabe Pharma’s ZYNLONTA® Hits Key Endpoint in Japanese r/r DLBCL Tr... Tanabe Pharma’s ZYNLONTA® Hits Key Endpoint in Japanese r/r DLBCL Tr... Read Post »
Clinical TrailsFDA Grants Class II Review for Unicycive’s Resubmission of OLC in Hyperph... FDA Grants Class II Review for Unicycive’s Resubmission of OLC in Hyperph... Read Post »
Drugs Safety AlertFDA Halts REGENXBIO’s RGX-111 and RGX-121 After CNS Tumor Case FDA Halts REGENXBIO’s RGX-111 and RGX-121 After CNS Tumor Case Read Post »
Clinical TrailsChugai Files Tecentriq for MRD-Positive Bladder Cancer in Japan Chugai Files Tecentriq for MRD-Positive Bladder Cancer in Japan Read Post »
Clinical TrailsBiogen’s Litifilimab Wins FDA Breakthrough Therapy Designation for Cutane... Biogen’s Litifilimab Wins FDA Breakthrough Therapy Designation for Cutane... Read Post »
Clinical TrailsFDA Grants Priority Review to Otsuka’s Centanafadine NDA for ADHD Across ... FDA Grants Priority Review to Otsuka’s Centanafadine NDA for ADHD Across ... Read Post »
New Drug ApprovalFDA Approves JnJ’s DARZALEX FASPRO Quadruplet for NDMM FDA Approves JnJ’s DARZALEX FASPRO Quadruplet for NDMM Read Post »
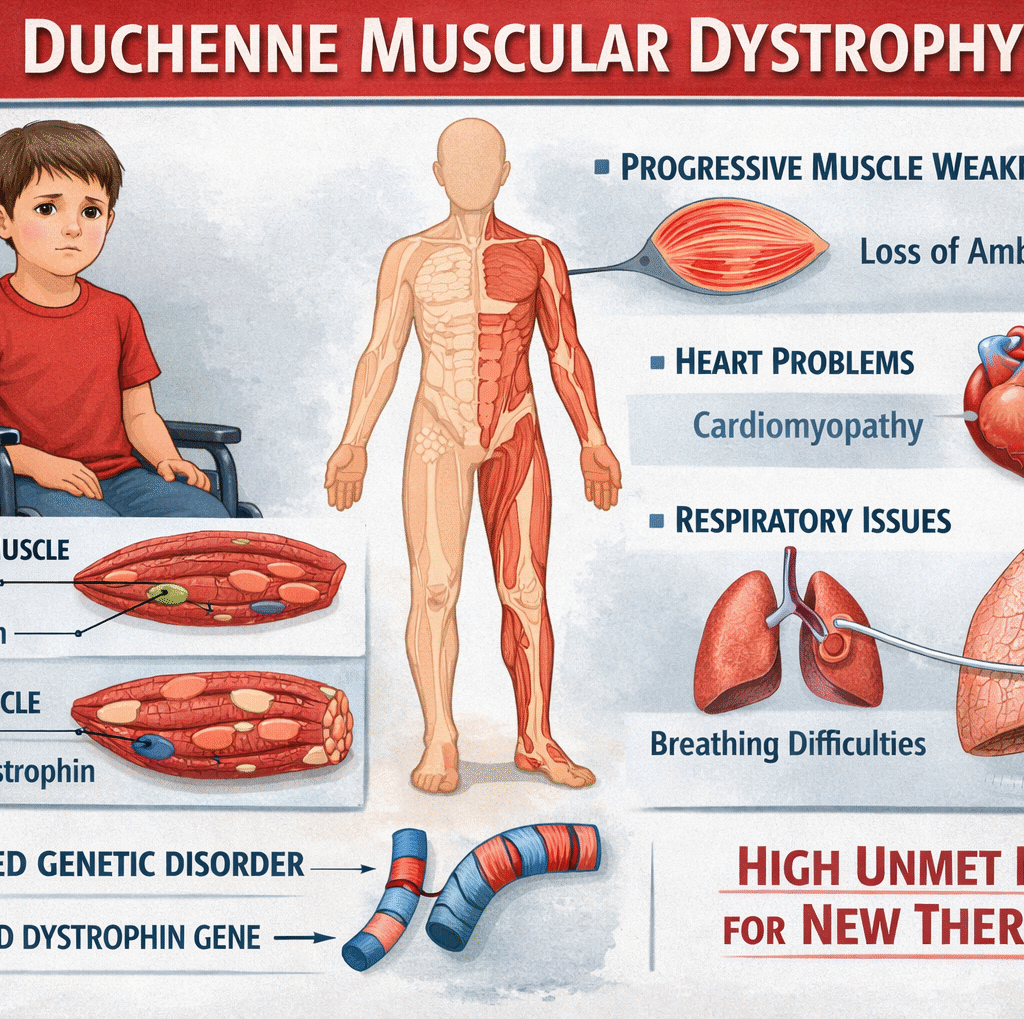
Clinical TrailsELEVIDYS Delivers 3-Year Sustained Motor Benefits in Ambulatory Duchenne Pa... ELEVIDYS Delivers 3-Year Sustained Motor Benefits in Ambulatory Duchenne Pa... Read Post »
Drugs Safety AlertFDA Lifts Clinical Hold on Intellia’s MAGNITUDE-2 Phase 3 Trial of Nex-z ... FDA Lifts Clinical Hold on Intellia’s MAGNITUDE-2 Phase 3 Trial of Nex-z ... Read Post »
Clinical TrailsNeurocrine Biosciences Launches Phase 2 Trial of NBI-1065890 for Tardive Dy... Neurocrine Biosciences Launches Phase 2 Trial of NBI-1065890 for Tardive Dy... Read Post »
Clinical TrailsRoche’s CT-388 Shows Up to 22.5% Placebo-Adjusted Weight Loss in Phase II... Roche’s CT-388 Shows Up to 22.5% Placebo-Adjusted Weight Loss in Phase II... Read Post »
New Drug ApprovalEU Expands GSK’s Arexvy Approval to All Adults Aged 18 and Older EU Expands GSK’s Arexvy Approval to All Adults Aged 18 and Older Read Post »
Clinical TrailsFDA Accepts Eisai-Biogen’s LEQEMBI IQLIK sBLA Under Priority Review FDA Accepts Eisai-Biogen’s LEQEMBI IQLIK sBLA Under Priority Review Read Post »
Food ResearchBest Diet While on GLP-1 Weight Loss Drugs (Ozempic, Wegovy, Zepbound): Evi... Best Diet While on GLP-1 Weight Loss Drugs (Ozempic, Wegovy, Zepbound): Evi... Read Post »
ResearchClosed-Loop Insulin Delivery Systems: The New Standard Closed-Loop Insulin Delivery Systems: The New Standard Read Post »
Clinical TrailsBausch Health’s RED-C Program Comes Up Short in Phase 3 Bausch Health’s RED-C Program Comes Up Short in Phase 3 Read Post »
Clinical TrailsAbbott Shares Five-Year Data Showing Neuromodulation Therapy Cuts Pain-Rela... Abbott Shares Five-Year Data Showing Neuromodulation Therapy Cuts Pain-Rela... Read Post »
Clinical TrailsSanofi’s Amlitelimab Strengthens Phase 3 Evidence in Atopic Dermatitis wi... Sanofi’s Amlitelimab Strengthens Phase 3 Evidence in Atopic Dermatitis wi... Read Post »
New Drug ApprovalSun Pharma Wins DCGI Approval for Noveltreat, Generic Semaglutide Injection... Sun Pharma Wins DCGI Approval for Noveltreat, Generic Semaglutide Injection... Read Post »
New Drug ApprovalGSK’s Trelegy Ellipta Approved by NMPA in China for Adult Asthma GSK’s Trelegy Ellipta Approved by NMPA in China for Adult Asthma Read Post »
Clinical TrailsCorcept Reports Overall Survival Benefit with Relacorilant in Platinum-Resi... Corcept Reports Overall Survival Benefit with Relacorilant in Platinum-Resi... Read Post »
Clinical TrailsBayer and BlueRock’s OpCT-001 Receives FDA Orphan Drug Designation for Re... Bayer and BlueRock’s OpCT-001 Receives FDA Orphan Drug Designation for Re... Read Post »
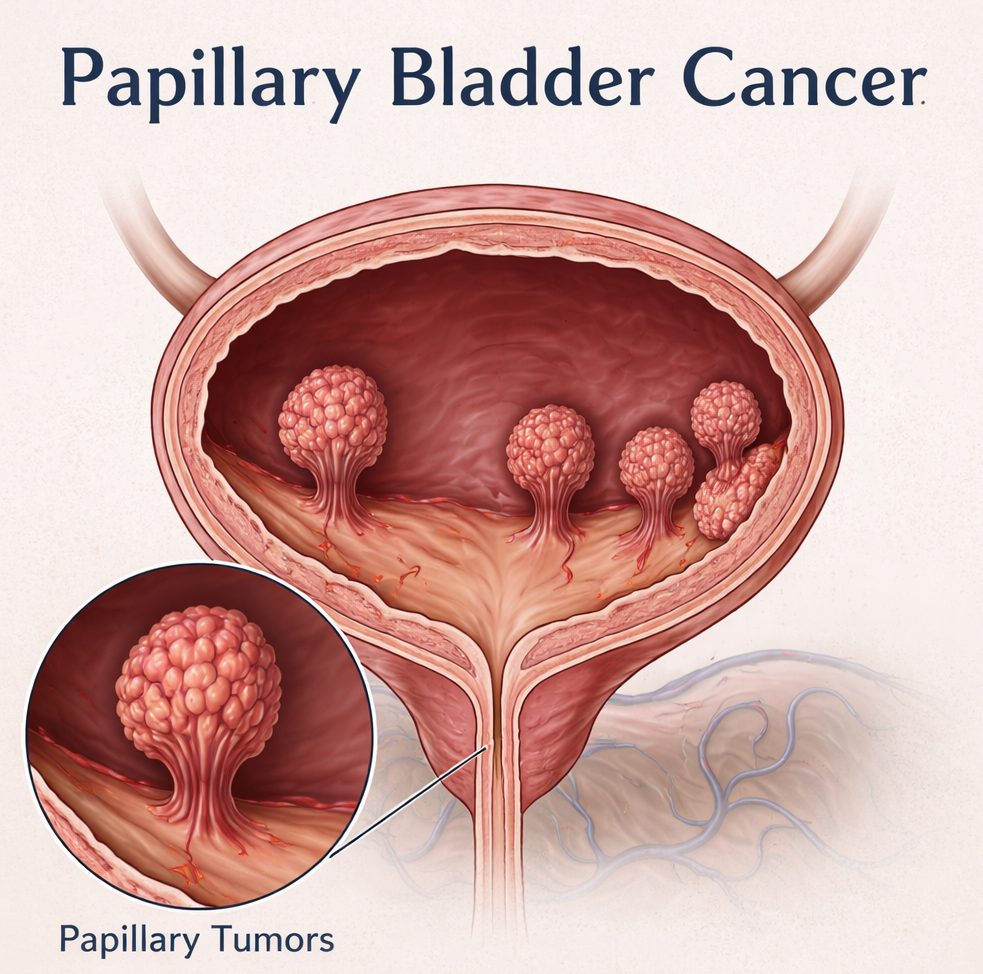
Clinical TrailsImmunityBio Advances ANKTIVA Toward FDA Resubmission in Papillary NMIBC ImmunityBio Advances ANKTIVA Toward FDA Resubmission in Papillary NMIBC Read Post »
Clinical TrailsFDA Grants Fast Track Designation to BioNTech’s BNT113 for HPV16-Positive... FDA Grants Fast Track Designation to BioNTech’s BNT113 for HPV16-Positive... Read Post »
Clinical TrailsFDA Grants Breakthrough Therapy Designation to Lilly’s Sofetabart Mipitec... FDA Grants Breakthrough Therapy Designation to Lilly’s Sofetabart Mipitec... Read Post »
Clinical TrailsModerna–Merck mRNA Cancer Vaccine Combo Shows Durable Five-Year Recurrenc... Moderna–Merck mRNA Cancer Vaccine Combo Shows Durable Five-Year Recurrenc... Read Post »
Drugs Safety AlertOrganon’s NEXPLANON Now Approved for Up to Five Years of Contraception wi... Organon’s NEXPLANON Now Approved for Up to Five Years of Contraception wi... Read Post »
New Drug ApprovalEMA Validates ENHERTU Plus Pertuzumab for First-Line HER2 Positive Metastat... EMA Validates ENHERTU Plus Pertuzumab for First-Line HER2 Positive Metastat... Read Post »
Clinical TrailsSimcere and Lynk Report Positive Phase III Data for Zemprocitinib in Modera... Simcere and Lynk Report Positive Phase III Data for Zemprocitinib in Modera... Read Post »
New Drug ApprovalNMPA Review Begins for Envafolimab NDA in Biliary Cancer NMPA Review Begins for Envafolimab NDA in Biliary Cancer Read Post »
Drugs Safety Alert Health TidingsFDA Issues Warning Letter to Telangana’s Palamur Biosciences Over GLP Ris... FDA Issues Warning Letter to Telangana’s Palamur Biosciences Over GLP Ris... Read Post »
Lifestyle Research WellnessSmall Steps, Longer Lives: Lancet Study Links Minutes of Movement to Lower ... Small Steps, Longer Lives: Lancet Study Links Minutes of Movement to Lower ... Read Post »
New Drug ApprovalEU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... EU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... Read Post »
Clinical TrailsAxsome Therapeutics Doses First Patient in Phase 3 FORWARD Trial of AXS-14 ... Axsome Therapeutics Doses First Patient in Phase 3 FORWARD Trial of AXS-14 ... Read Post »
Clinical TrailsJohnson & Johnson Presents New Evidence That CAPLYTA With Antidepressan... Johnson & Johnson Presents New Evidence That CAPLYTA With Antidepressan... Read Post »
New Drug ApprovalAtossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Atossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Read Post »
New Drug ApprovalNovartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Novartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Read Post »
Clinical TrailsAbbVie and Genmab Report Phase 3 Results for Epcoritamab in Relapsed DLBCL AbbVie and Genmab Report Phase 3 Results for Epcoritamab in Relapsed DLBCL Read Post »
Clinical TrailsTeva’s AJOVY Demonstrates Strong Efficacy in Reducing Pediatric Migraine ... Teva’s AJOVY Demonstrates Strong Efficacy in Reducing Pediatric Migraine ... Read Post »
New Drug ApprovalChina NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... China NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... Read Post »
ResearchINGREZZA Shows Nearly Double VMAT2 Target Occupancy Compared to AUSTEDO XR ... INGREZZA Shows Nearly Double VMAT2 Target Occupancy Compared to AUSTEDO XR ... Read Post »
Clinical TrailsTanabe Pharma Reports Positive Phase 3 INSPIRE Results for MT-7117 in EPP a... Tanabe Pharma Reports Positive Phase 3 INSPIRE Results for MT-7117 in EPP a... Read Post »
Clinical TrailsPhase 3 MajesTEC-9 Trial Shows TECVAYLI® Monotherapy Improves Survival in ... Phase 3 MajesTEC-9 Trial Shows TECVAYLI® Monotherapy Improves Survival in ... Read Post »
New Drug ApprovalAbbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Abbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Read Post »
New Drug ApprovalSebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Sebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Read Post »
Drugs Safety AlertFDA Removes Suicidal Ideation Warning From GLP-1 Weight-Loss Drugs FDA Removes Suicidal Ideation Warning From GLP-1 Weight-Loss Drugs Read Post »
Clinical TrailsBristol Myers Squibb Reports Positive Phase 3 SCOUT-HCM Results for Camzyos... Bristol Myers Squibb Reports Positive Phase 3 SCOUT-HCM Results for Camzyos... Read Post »
New Drug ApprovalBiogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Biogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Read Post »