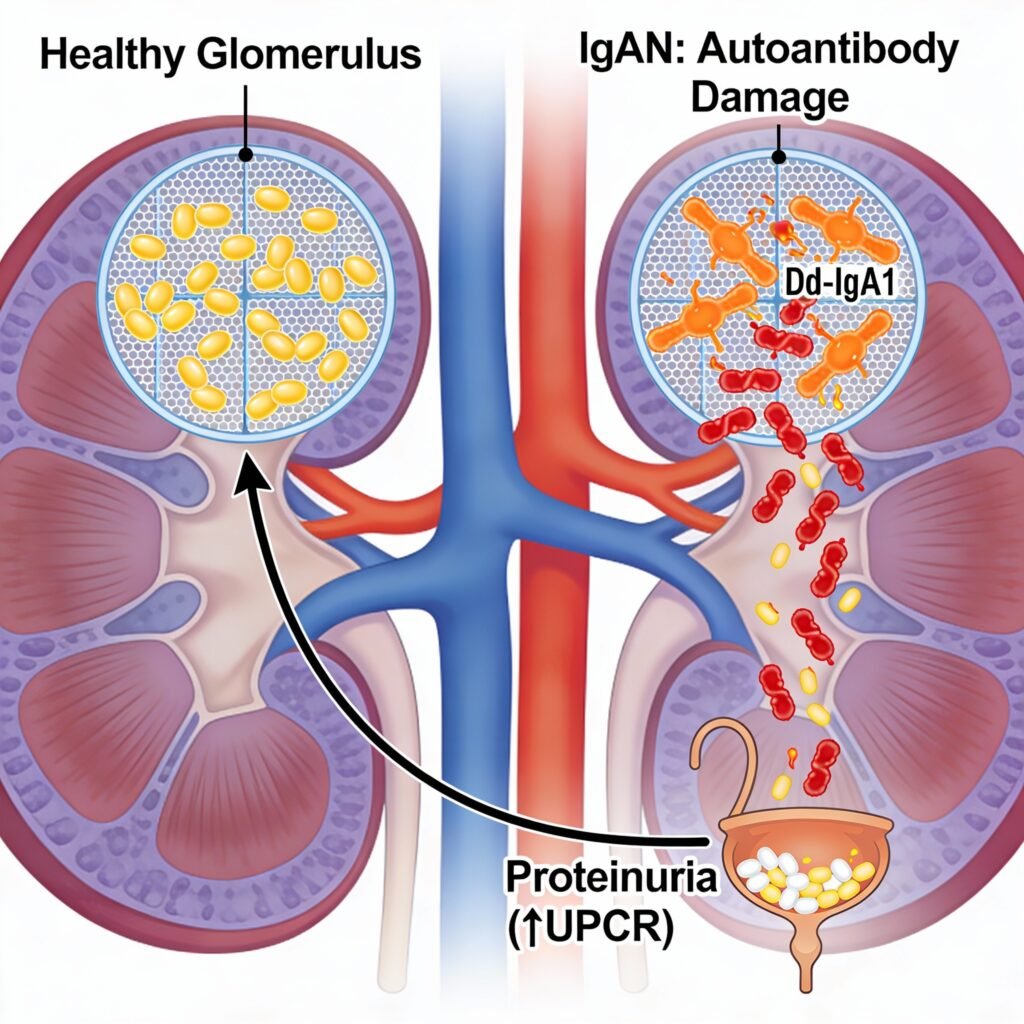
New Drug ApprovalFDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... FDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... Read Post »
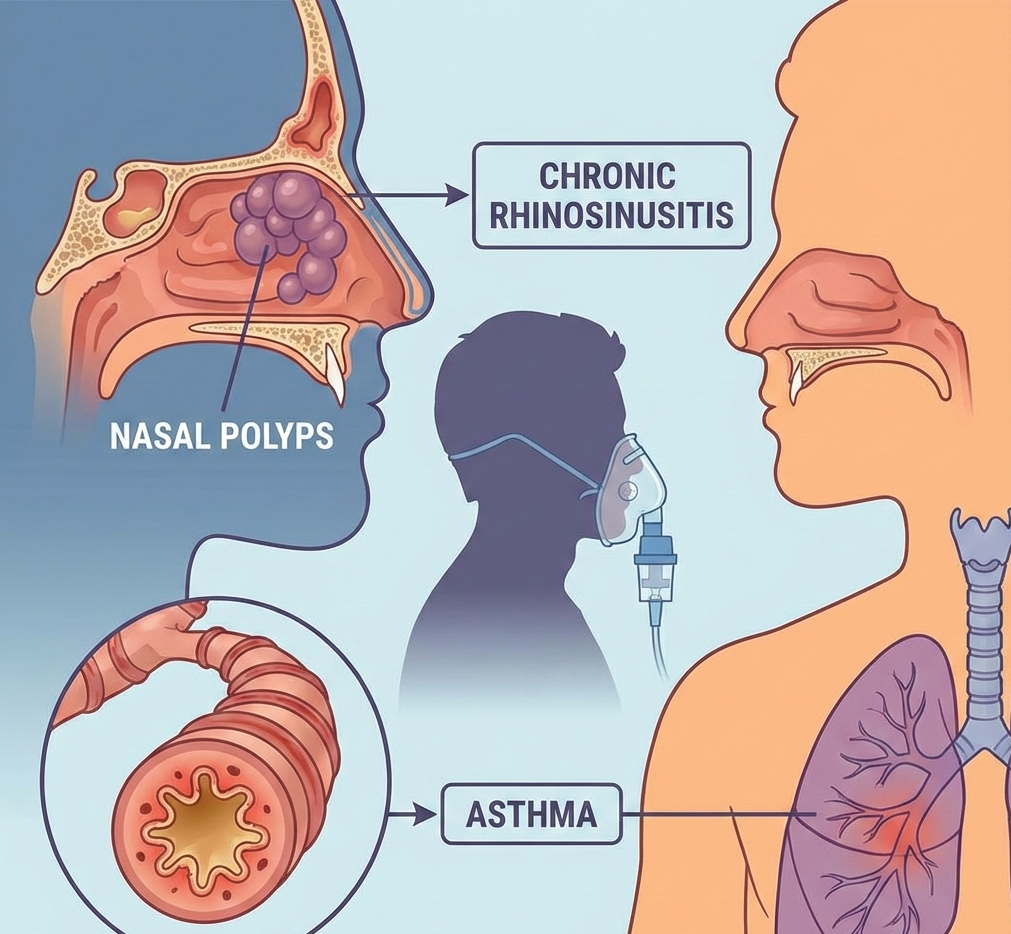
New Drug ApprovalJapan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Japan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Read Post »
New Drug ApprovalZai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Zai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Read Post »
New Drug ApprovalChina Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s China Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s Read Post »
New Drug ApprovalHealth Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Health Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Read Post »
New Drug ApprovalModerna Files With FDA, EMA, Health Canada and TGA for New mRNA Flu Shot Moderna Files With FDA, EMA, Health Canada and TGA for New mRNA Flu Shot Read Post »
New Drug ApprovalAltimmune’s Pemvidutide Granted FDA Breakthrough Therapy Designation for ... Altimmune’s Pemvidutide Granted FDA Breakthrough Therapy Designation for ... Read Post »
New Drug ApprovalGSK’s Nucala (Mepolizumab) Gains China NMPA Approval for Eosinophilic... GSK’s Nucala (Mepolizumab) Gains China NMPA Approval for Eosinophilic... Read Post »
New Drug ApprovalSanofi’s Tzield (Teplizumab) Gains FDA Priority Review for 1-Year-Old... Sanofi’s Tzield (Teplizumab) Gains FDA Priority Review for 1-Year-Old... Read Post »
New Drug ApprovalMHRA Approves Kostaive (Zapomeran) sa-mRNA COVID Booster for UK Adults-Key ... MHRA Approves Kostaive (Zapomeran) sa-mRNA COVID Booster for UK Adults-Key ... Read Post »
New Drug ApprovalUltragenyx Completes Rolling BLA for First-in-Class DTX401 Gene Therapy in ... Ultragenyx Completes Rolling BLA for First-in-Class DTX401 Gene Therapy in ... Read Post »
New Drug ApprovalFDA Accepts Axsome’s AXS-05 Supplemental NDA and Grants Priority Review f... FDA Accepts Axsome’s AXS-05 Supplemental NDA and Grants Priority Review f... Read Post »
New Drug ApprovalFDA Clears NEREUS, First New Motion Sickness Drug in Decades FDA Clears NEREUS, First New Motion Sickness Drug in Decades Read Post »
New Drug ApprovalUnicycive Resubmits NDA for Oxylanthanum Carbonate for Hyperphosphatemia Ma... Unicycive Resubmits NDA for Oxylanthanum Carbonate for Hyperphosphatemia Ma... Read Post »
New Drug ApprovalFDA Grants Breakthrough Therapy to Ulixacaltamide (PRAX-944) for Essential ... FDA Grants Breakthrough Therapy to Ulixacaltamide (PRAX-944) for Essential ... Read Post »
New Drug ApprovalFirst-in-Class TRPV1 Inhibitor Avarept Approved in Japan for Dry Eye Diseas... First-in-Class TRPV1 Inhibitor Avarept Approved in Japan for Dry Eye Diseas... Read Post »
New Drug Approval ResearchGrifols’ FESILTY (Fibrinogen): Evidence, Safety, and Clinical Use Explain... Grifols’ FESILTY (Fibrinogen): Evidence, Safety, and Clinical Use Explain... Read Post »
New Drug ApprovalENHERTU Approved in China for HER2-Low and HER2-Ultralow Metastatic Breast ... ENHERTU Approved in China for HER2-Low and HER2-Ultralow Metastatic Breast ... Read Post »
New Drug ApprovalFDA Approves YARTEMLEA: First TA-TMA Therapy Post-Stem Cell Transplant FDA Approves YARTEMLEA: First TA-TMA Therapy Post-Stem Cell Transplant Read Post »
New Drug ApprovalIncyte’s Zynyz Receives First Approval in Japan for Advanced SCAC Incyte’s Zynyz Receives First Approval in Japan for Advanced SCAC Read Post »
New Drug ApprovalJapan Expands Pediatric Asthma Treatment with Dupixent Approval Japan Expands Pediatric Asthma Treatment with Dupixent Approval Read Post »
New Drug ApprovalZai Lab Wins China Approval for COBENFY, a First-in-Class Muscarinic Therap... Zai Lab Wins China Approval for COBENFY, a First-in-Class Muscarinic Therap... Read Post »
New Drug ApprovalU.S. FDA Approves Agios’ AQVESME (Mitapivat) as First Treatment for Anemi... U.S. FDA Approves Agios’ AQVESME (Mitapivat) as First Treatment for Anemi... Read Post »
New Drug ApprovalJapan Approves Finerenone for Chronic Heart Failure with Preserved or Mildl... Japan Approves Finerenone for Chronic Heart Failure with Preserved or Mildl... Read Post »
New Drug ApprovalEuropean Commission Approves TREMFYA® (guselkumab) for Paediatric Plaque P... European Commission Approves TREMFYA® (guselkumab) for Paediatric Plaque P... Read Post »
New Drug ApprovalNovo Nordisk’s Wegovy Pill Wins FDA Approval Following OASIS Trial Result... Novo Nordisk’s Wegovy Pill Wins FDA Approval Following OASIS Trial Result... Read Post »
New Drug ApprovalSubcutaneous Lunsumio VELO™ Approved: 75% ORR in Heavily Pretreated Folli... Subcutaneous Lunsumio VELO™ Approved: 75% ORR in Heavily Pretreated Folli... Read Post »
New Drug ApprovalMYQORZO Aficamten FDA Approval: Breakthrough for Symptomatic oHCM Treatment... MYQORZO Aficamten FDA Approval: Breakthrough for Symptomatic oHCM Treatment... Read Post »
New Drug ApprovalFDA Approves Expanded Indication for Johnson & Johnson’s TRUFILL n‑... FDA Approves Expanded Indication for Johnson & Johnson’s TRUFILL n‑... Read Post »
New Drug ApprovalNovo Nordisk Pushes Obesity Innovation Forward With FDA Filing for CagriSem... Novo Nordisk Pushes Obesity Innovation Forward With FDA Filing for CagriSem... Read Post »
New Drug ApprovalinMIND Trial Success: Minjuvi (Tafasitamab) Now EU-Approved for Relapsed Fo... inMIND Trial Success: Minjuvi (Tafasitamab) Now EU-Approved for Relapsed Fo... Read Post »
New Drug ApprovalSaphnelo Gains EU Approval for At-Home Subcutaneous Self-Administration in ... Saphnelo Gains EU Approval for At-Home Subcutaneous Self-Administration in ... Read Post »
New Drug ApprovalAddyi FDA Approval 2025: Now for Postmenopausal Women with Low Libido Addyi FDA Approval 2025: Now for Postmenopausal Women with Low Libido Read Post »
New Drug ApprovalBreakthrough PCSK9 Inhibitor LEROCHOL Approved for Hypercholesterolemia Breakthrough PCSK9 Inhibitor LEROCHOL Approved for Hypercholesterolemia Read Post »
New Drug ApprovalExdensur (Depemokimab) Secures First Global Approval in the UK, Followed by... Exdensur (Depemokimab) Secures First Global Approval in the UK, Followed by... Read Post »
New Drug ApprovalFDA Approves CARDAMYST™ (Etripamil) as First Self-Administered Nasal Spra... FDA Approves CARDAMYST™ (Etripamil) as First Self-Administered Nasal Spra... Read Post »
New Drug ApprovalNew FDA Approval: Uplizna for gMG in Adults New FDA Approval: Uplizna for gMG in Adults Read Post »
New Drug ApprovalEMA CHMP December 2025 Meeting: 7 New Medicine Approvals and 12 Indication ... EMA CHMP December 2025 Meeting: 7 New Medicine Approvals and 12 Indication ... Read Post »
New Drug ApprovalFDA Approves Nuzolvence (Zoliflodacin): First-in-Class Oral Antibiotic for ... FDA Approves Nuzolvence (Zoliflodacin): First-in-Class Oral Antibiotic for ... Read Post »
New Drug ApprovalFDA Approves First Cell-Based Therapy Waskyra for Rare WAS Immunodeficiency FDA Approves First Cell-Based Therapy Waskyra for Rare WAS Immunodeficiency Read Post »
New Drug ApprovalFDA’s First CNPV Approval: Augmentin XR Goes Domestic FDA’s First CNPV Approval: Augmentin XR Goes Domestic Read Post »
New Drug ApprovalFDA Qualifies First AI Tool under Drug Development Tool Program, Paving the... FDA Qualifies First AI Tool under Drug Development Tool Program, Paving the... Read Post »
New Drug ApprovalFDA Approves Omisirge for Severe Aplastic Anemia: A New Option for Patients... FDA Approves Omisirge for Severe Aplastic Anemia: A New Option for Patients... Read Post »
New Drug ApprovalFDA Approves Breyanzi as First CAR T-Cell Therapy for Relapsed/Refractory M... FDA Approves Breyanzi as First CAR T-Cell Therapy for Relapsed/Refractory M... Read Post »
Clinical Trails New Drug ApprovalIonis’ Zilganersen Earns FDA Breakthrough Therapy Designation for Ultra R... Ionis’ Zilganersen Earns FDA Breakthrough Therapy Designation for Ultra R... Read Post »
New Drug ApprovalSun Pharma Launches Ilumya® in India: A Global Innovative Drug for Moderat... Sun Pharma Launches Ilumya® in India: A Global Innovative Drug for Moderat... Read Post »
New Drug ApprovalBaxdrostat NDA Accepted Under FDA Priority Review for Hard-to-Control High ... Baxdrostat NDA Accepted Under FDA Priority Review for Hard-to-Control High ... Read Post »
New Drug ApprovalAmneal Secure FDA Nod for Albuterol Inhaler and Cyclosporine Eye Drops in E... Amneal Secure FDA Nod for Albuterol Inhaler and Cyclosporine Eye Drops in E... Read Post »
New Drug ApprovalUS FDA Accepts Wockhardt’s NDA for Zaynich, Marking a Historic First for ... US FDA Accepts Wockhardt’s NDA for Zaynich, Marking a Historic First for ... Read Post »
New Drug ApprovalChina’s First Domestic IL-23p19 Antibody PECONDLE Gains NMPA Approval... China’s First Domestic IL-23p19 Antibody PECONDLE Gains NMPA Approval... Read Post »