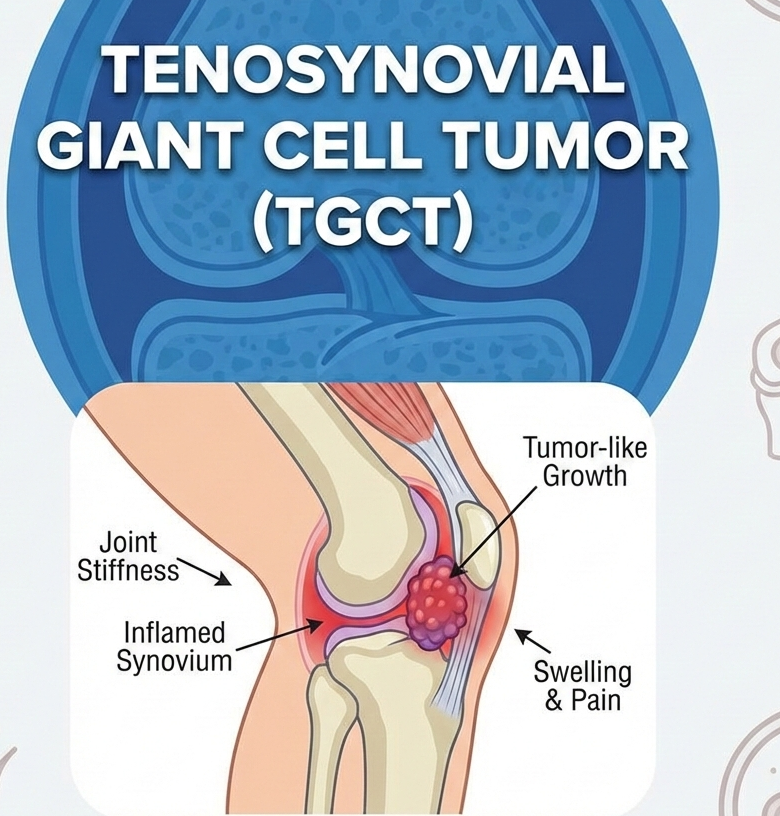
New Drug ApprovalHERNEXEOS® Gains FDA Nod as First-Line Option for HER2-Mutant NSCLC HERNEXEOS® Gains FDA Nod as First-Line Option for HER2-Mutant NSCLC Read Post »
New Drug ApprovalFDA Approves Once-Weekly YUVIWEL® for Pediatric Achondroplasia FDA Approves Once-Weekly YUVIWEL® for Pediatric Achondroplasia Read Post »
New Drug ApprovalFDA Approves BioMarin PALYNZIQ for Adolescents with PKU FDA Approves BioMarin PALYNZIQ for Adolescents with PKU Read Post »
New Drug ApprovalJ&J Seeks FDA Approval of IMAAVY® as First Treatment for wAIHA J&J Seeks FDA Approval of IMAAVY® as First Treatment for wAIHA Read Post »
New Drug ApprovalFDA Grants Full Approval to Pfizer’s BRAFTOVI Regimen FDA Grants Full Approval to Pfizer’s BRAFTOVI Regimen Read Post »
New Drug ApprovalDupixent Approved by FDA: Sanofi & Regeneron’s First Treatment fo... Dupixent Approved by FDA: Sanofi & Regeneron’s First Treatment fo... Read Post »
New Drug ApprovalFDA Approves Zepbound 4-Dose KwikPen for Obesity Care FDA Approves Zepbound 4-Dose KwikPen for Obesity Care Read Post »
New Drug ApprovalFDA Approves BYSANTI™: A New Ally in Bipolar I and Schizophrenia Treatmen... FDA Approves BYSANTI™: A New Ally in Bipolar I and Schizophrenia Treatmen... Read Post »
New Drug ApprovalChugai report Anti-CD20 Monoclonal Antibody Rituxan® Approved for AIHA Chugai report Anti-CD20 Monoclonal Antibody Rituxan® Approved for AIHA Read Post »
New Drug ApprovalAbbVie’s VENCLEXTA Taps AZ’s Acalabrutinib for FDA’s Firs... AbbVie’s VENCLEXTA Taps AZ’s Acalabrutinib for FDA’s Firs... Read Post »
New Drug ApprovalEuropean Commission Grants Conditional Approval for ANKTIVA® in BCG-Unresp... European Commission Grants Conditional Approval for ANKTIVA® in BCG-Unresp... Read Post »
New Drug ApprovalFDA Approves Simplified Monthly Dosing for RYBREVANT FASPRO™ in EGFR-Muta... FDA Approves Simplified Monthly Dosing for RYBREVANT FASPRO™ in EGFR-Muta... Read Post »
New Drug ApprovalGSK’s Exdensur Gains EU Approval for Severe Asthma and CRSwNP GSK’s Exdensur Gains EU Approval for Severe Asthma and CRSwNP Read Post »
New Drug ApprovalAmgen’s UPLIZNA® Approved for gMG in EU Amgen’s UPLIZNA® Approved for gMG in EU Read Post »
New Drug ApprovalKEYTRUDA® (pembrolizumab) and KEYTRUDA QLEX™ Approved in Combination The... KEYTRUDA® (pembrolizumab) and KEYTRUDA QLEX™ Approved in Combination The... Read Post »
New Drug ApprovalNovocure’s Optune Pax Receives FDA Approval in Pancreatic Cancer Novocure’s Optune Pax Receives FDA Approval in Pancreatic Cancer Read Post »
New Drug ApprovalGSK’s Nucala Secures European Approval for Eosinophilic COPD GSK’s Nucala Secures European Approval for Eosinophilic COPD Read Post »
New Drug ApprovalFDA Approves VYBRIQUE as First Oral Film for Erectile Dysfunction FDA Approves VYBRIQUE as First Oral Film for Erectile Dysfunction Read Post »
New Drug ApprovalAbbVie Seeks FDA, EMA Approval of RINVOQ in Vitiligo AbbVie Seeks FDA, EMA Approval of RINVOQ in Vitiligo Read Post »
New Drug ApprovalBayer’s Nubeqa Approved for mHSPC in China Bayer’s Nubeqa Approved for mHSPC in China Read Post »
New Drug ApprovalImfinzi Boosts Survival in Gastric Cancer, Gains CHMP Endorsement Imfinzi Boosts Survival in Gastric Cancer, Gains CHMP Endorsement Read Post »
New Drug ApprovalCHMP Backs Rezurock for Chronic GVHD After Re-Examination CHMP Backs Rezurock for Chronic GVHD After Re-Examination Read Post »
New Drug ApprovalIncyte’s Zynyz Wins Positive CHMP Opinion in Advanced Anal Cancer Incyte’s Zynyz Wins Positive CHMP Opinion in Advanced Anal Cancer Read Post »
New Drug ApprovalEMA CHMP Recommends Bayer’s Finerenone for Heart Failure with LVEF �... EMA CHMP Recommends Bayer’s Finerenone for Heart Failure with LVEF �... Read Post »
New Drug ApprovalJ&J Scores CHMP Nod for AKEEGA in High-Risk Prostate Cancer J&J Scores CHMP Nod for AKEEGA in High-Risk Prostate Cancer Read Post »
New Drug ApprovalFDA Approves Tenpoint’s YUVEZZI Eye Drop for Presbyopia Treatment FDA Approves Tenpoint’s YUVEZZI Eye Drop for Presbyopia Treatment Read Post »
New Drug ApprovalFDA Approves JnJ’s DARZALEX FASPRO Quadruplet for NDMM FDA Approves JnJ’s DARZALEX FASPRO Quadruplet for NDMM Read Post »
New Drug ApprovalEU Expands GSK’s Arexvy Approval to All Adults Aged 18 and Older EU Expands GSK’s Arexvy Approval to All Adults Aged 18 and Older Read Post »
New Drug ApprovalSun Pharma Wins DCGI Approval for Noveltreat, Generic Semaglutide Injection... Sun Pharma Wins DCGI Approval for Noveltreat, Generic Semaglutide Injection... Read Post »
New Drug ApprovalGSK’s Trelegy Ellipta Approved by NMPA in China for Adult Asthma GSK’s Trelegy Ellipta Approved by NMPA in China for Adult Asthma Read Post »
New Drug ApprovalEMA Validates ENHERTU Plus Pertuzumab for First-Line HER2 Positive Metastat... EMA Validates ENHERTU Plus Pertuzumab for First-Line HER2 Positive Metastat... Read Post »
New Drug ApprovalNMPA Review Begins for Envafolimab NDA in Biliary Cancer NMPA Review Begins for Envafolimab NDA in Biliary Cancer Read Post »
New Drug ApprovalEU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... EU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... Read Post »
New Drug ApprovalAtossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Atossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Read Post »
New Drug ApprovalNovartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Novartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Read Post »
New Drug ApprovalChina NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... China NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... Read Post »
New Drug ApprovalAbbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Abbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Read Post »
New Drug ApprovalSebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Sebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Read Post »
New Drug ApprovalBiogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Biogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Read Post »
New Drug ApprovalGSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix GSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix Read Post »
New Drug ApprovalFDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment FDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment Read Post »
New Drug ApprovalFDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... FDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... Read Post »
New Drug ApprovalFDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... FDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... Read Post »
New Drug ApprovalSanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Sanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Read Post »
New Drug ApprovalBayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Bayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Read Post »
New Drug ApprovalChina Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... China Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... Read Post »
New Drug ApprovalMilestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Milestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Read Post »
New Drug ApprovalFDA expands Cablivi approval to pediatric patients aged 12 and older with a... FDA expands Cablivi approval to pediatric patients aged 12 and older with a... Read Post »
New Drug ApprovalTakeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Takeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Read Post »