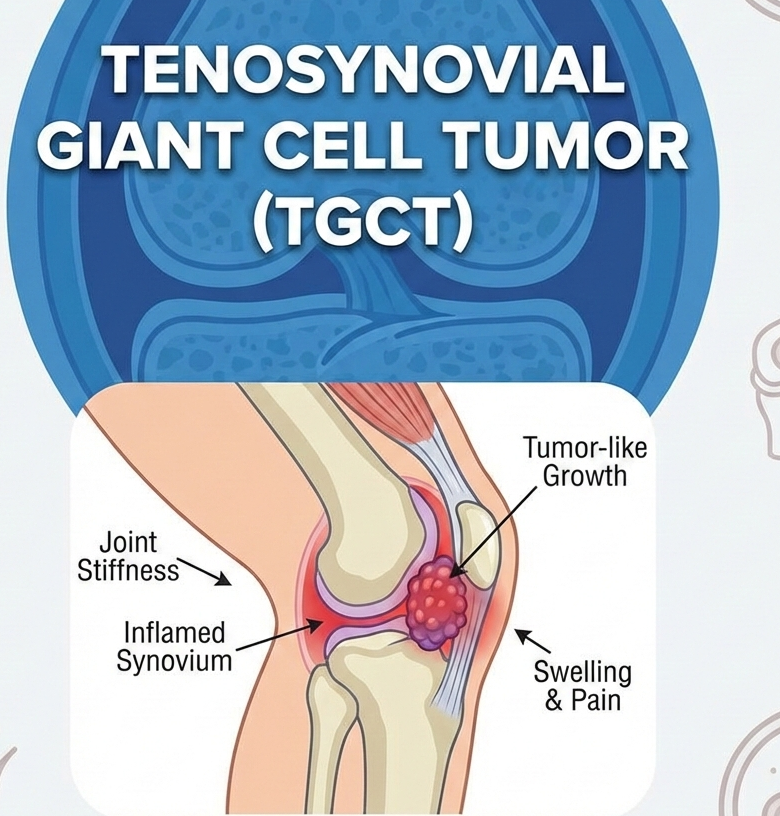
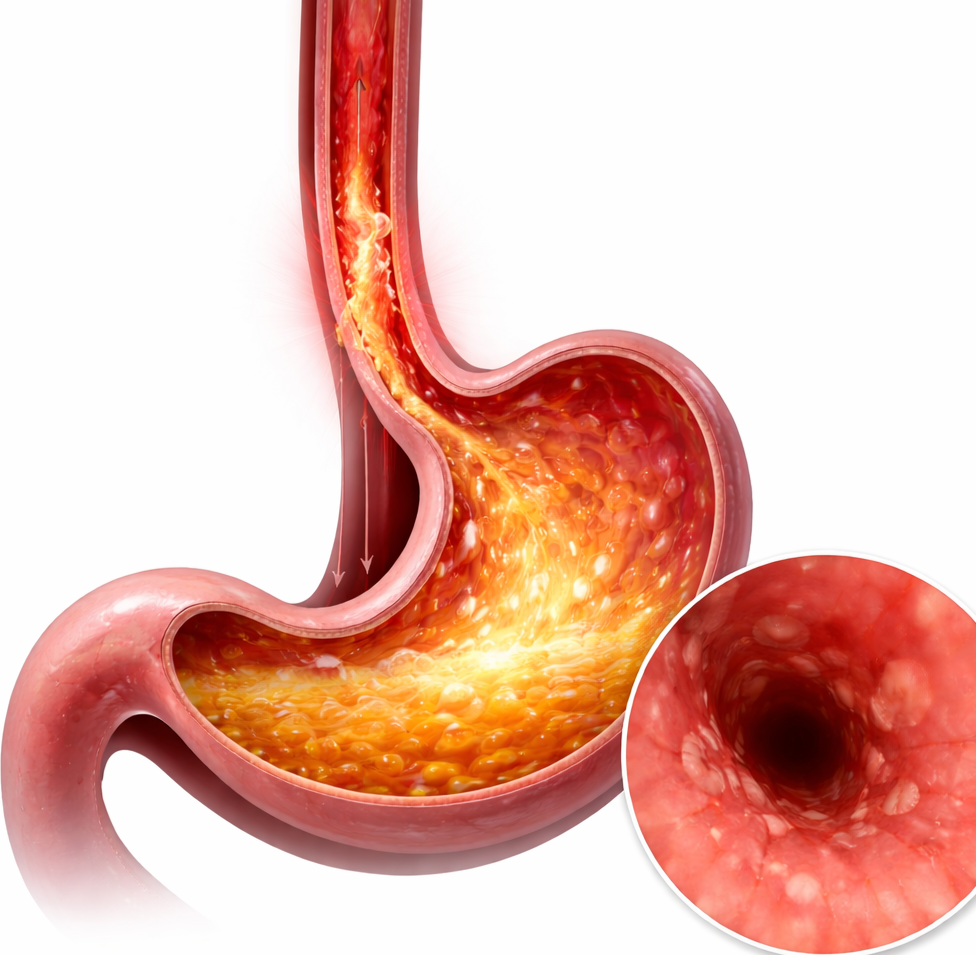
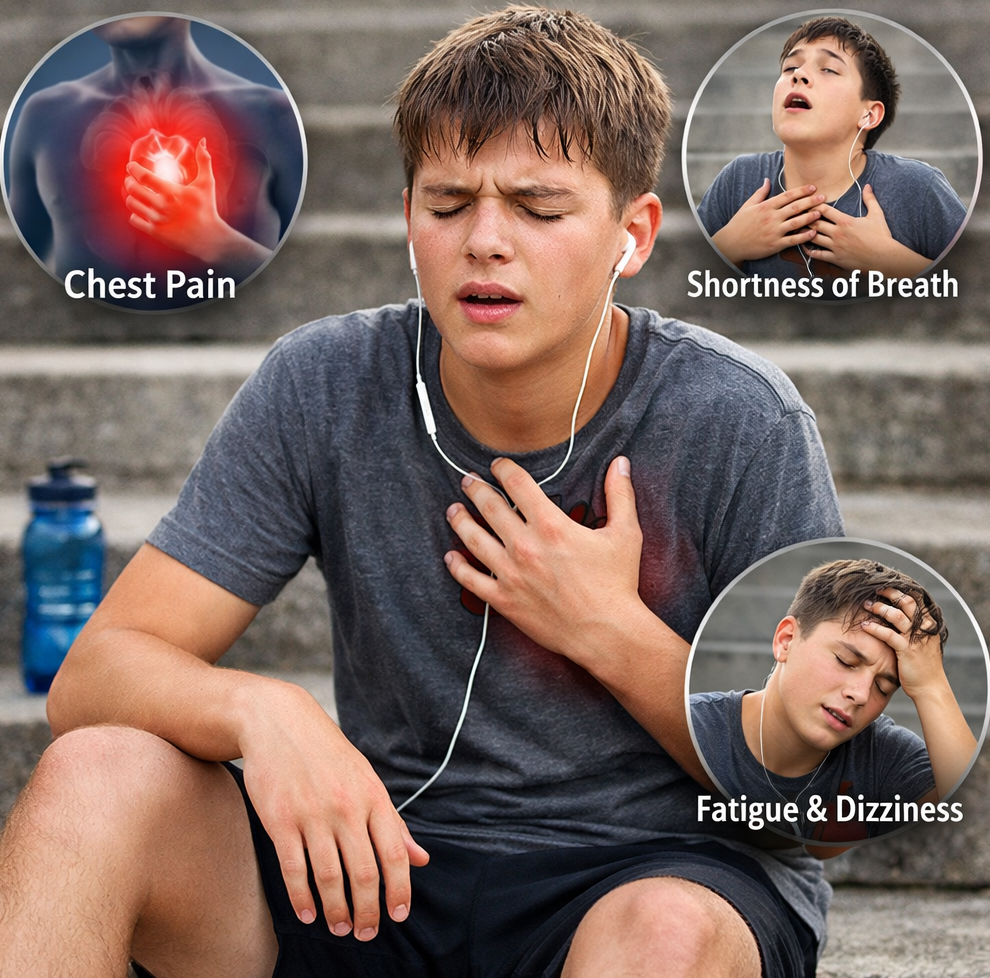
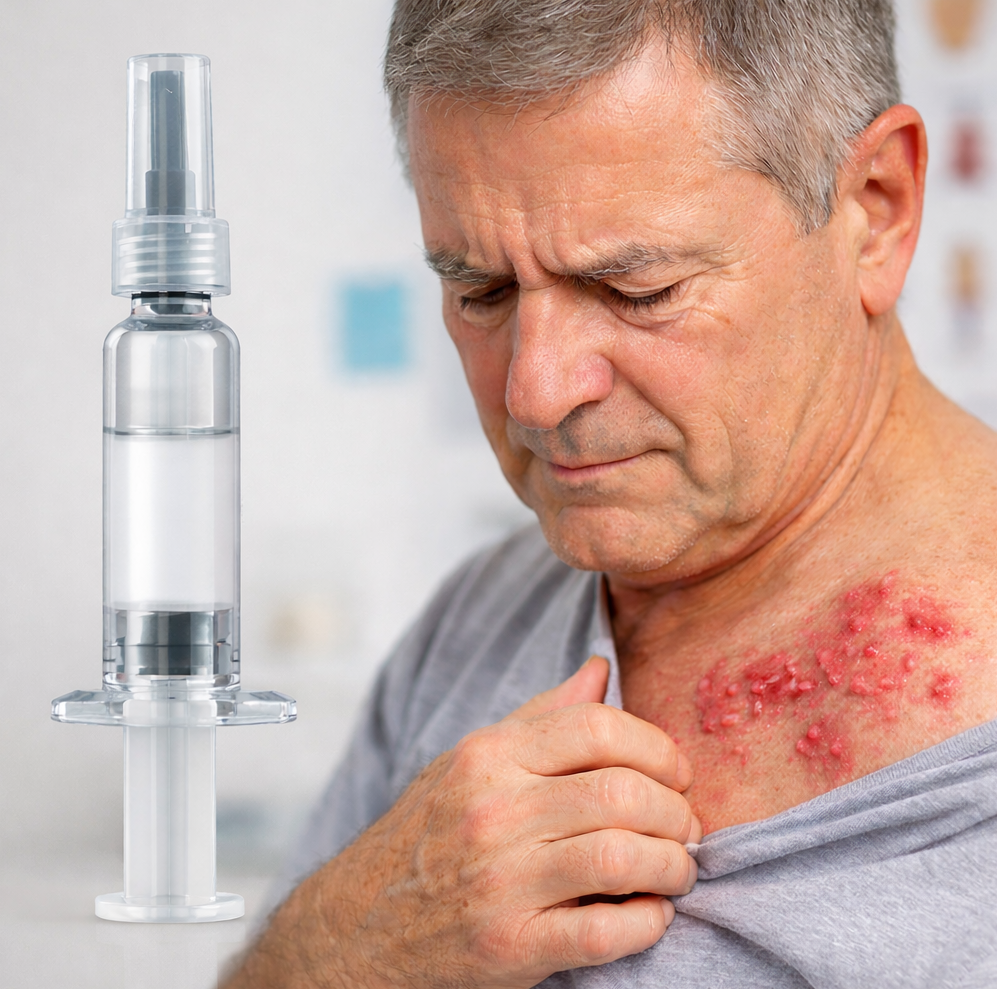
Food WellnessEat Well, Fast Well: Simple Nutrition Strategies for Ramadan Eat Well, Fast Well: Simple Nutrition Strategies for Ramadan Read Post »
New Drug ApprovalEU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... EU Approves Eylea 8 mg for Retinal Vein Occlusion After Durable Results in ... Read Post »
Clinical TrailsAxsome Therapeutics Doses First Patient in Phase 3 FORWARD Trial of AXS-14 ... Axsome Therapeutics Doses First Patient in Phase 3 FORWARD Trial of AXS-14 ... Read Post »
Clinical TrailsJohnson & Johnson Presents New Evidence That CAPLYTA With Antidepressan... Johnson & Johnson Presents New Evidence That CAPLYTA With Antidepressan... Read Post »
New Drug ApprovalAtossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Atossa Therapeutics Secures FDA Orphan Drug Designation for (Z)-Endoxifen i... Read Post »
New Drug ApprovalNovartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Novartis’ Ianalumab Secures FDA Breakthrough Therapy Designation for Sjö... Read Post »
Clinical TrailsAbbVie and Genmab Report Phase 3 Results for Epcoritamab in Relapsed DLBCL AbbVie and Genmab Report Phase 3 Results for Epcoritamab in Relapsed DLBCL Read Post »
Food WellnessHow Much Fiber Is Enough? The Truth About Fibermaxxing How Much Fiber Is Enough? The Truth About Fibermaxxing Read Post »
Clinical TrailsTeva’s AJOVY Demonstrates Strong Efficacy in Reducing Pediatric Migraine ... Teva’s AJOVY Demonstrates Strong Efficacy in Reducing Pediatric Migraine ... Read Post »
New Drug ApprovalChina NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... China NMPA Approves Two Sanofi-Licensed Innovative Therapies: Myqorzo (Afic... Read Post »
ResearchINGREZZA Shows Nearly Double VMAT2 Target Occupancy Compared to AUSTEDO XR ... INGREZZA Shows Nearly Double VMAT2 Target Occupancy Compared to AUSTEDO XR ... Read Post »
Clinical TrailsTanabe Pharma Reports Positive Phase 3 INSPIRE Results for MT-7117 in EPP a... Tanabe Pharma Reports Positive Phase 3 INSPIRE Results for MT-7117 in EPP a... Read Post »
Clinical TrailsPhase 3 MajesTEC-9 Trial Shows TECVAYLI® Monotherapy Improves Survival in ... Phase 3 MajesTEC-9 Trial Shows TECVAYLI® Monotherapy Improves Survival in ... Read Post »
Policy & AcquisitionsFDA, EMA Issue New Guidance on AI Use in Drug Development FDA, EMA Issue New Guidance on AI Use in Drug Development Read Post »
New Drug ApprovalAbbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Abbisko Therapeutics Achieves Major Milestone as FDA Accepts NDA for Pimico... Read Post »
New Drug ApprovalSebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Sebela Submits NDA for Tegoprazan as Potential First-in-Class P-CAB for GER... Read Post »
Drugs Safety AlertFDA Removes Suicidal Ideation Warning From GLP-1 Weight-Loss Drugs FDA Removes Suicidal Ideation Warning From GLP-1 Weight-Loss Drugs Read Post »
Policy & AcquisitionsNVIDIA and Eli Lilly Bet $1B on AI-Driven Drug Discovery NVIDIA and Eli Lilly Bet $1B on AI-Driven Drug Discovery Read Post »
Clinical TrailsBristol Myers Squibb Reports Positive Phase 3 SCOUT-HCM Results for Camzyos... Bristol Myers Squibb Reports Positive Phase 3 SCOUT-HCM Results for Camzyos... Read Post »
New Drug ApprovalBiogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Biogen’s High-Dose SPINRAZA Approved in EU Following DEVOTE Study Results Read Post »
New Drug ApprovalGSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix GSK Receives EU Approval for Ready-to-Use Prefilled Syringe Shingrix Read Post »
Health TidingsAquestive Therapeutics Reports FDA-Identified Deficiencies in Anaphylm NDA,... Aquestive Therapeutics Reports FDA-Identified Deficiencies in Anaphylm NDA,... Read Post »
New Drug ApprovalFDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment FDA Accepts Camurus NDA Resubmission for Oclaiz™ in Acromegaly Treatment Read Post »
New Drug ApprovalFDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... FDA Approves Zycubo (Copper Histidinate) Injection as First-Ever Treatment ... Read Post »
Health TidingsFDA Issues Complete Response Letter for Atara’s EBVALLO in EBV-Positive P... FDA Issues Complete Response Letter for Atara’s EBVALLO in EBV-Positive P... Read Post »
New Drug ApprovalFDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... FDA Grants Breakthrough Therapy Designation to Encoded Therapeutics’ ETX1... Read Post »
Clinical TrailsJohnson & Johnson’s RYBREVANT® Shows Promising Long-Term Results in ... Johnson & Johnson’s RYBREVANT® Shows Promising Long-Term Results in ... Read Post »
New Drug ApprovalSanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Sanofi’s Teizeild Approved in EU to Delay Stage 3 Type 1 Diabetes Onset i... Read Post »
Drugs Safety AlertFDA CBER Flags Rare Febrile Seizure Risk Linked to Influenza Vaccines FDA CBER Flags Rare Febrile Seizure Risk Linked to Influenza Vaccines Read Post »
Policy & AcquisitionsBayer and Soufflé Therapeutics Partner to Develop Heart-Targeted siRNA The... Bayer and Soufflé Therapeutics Partner to Develop Heart-Targeted siRNA The... Read Post »
New Drug ApprovalBayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Bayer’s Sevabertinib Gets Fast-Track Status in U.S. and China for HER2-Mu... Read Post »
New Drug ApprovalChina Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... China Grants First-in-World Approval to BeOne Medicines’ Sonrotoclax ... Read Post »
New Drug ApprovalMilestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Milestone’s Etripamil Nasal Spray Accepted for EMA Review in PSVT Read Post »
Clinical TrailsBepirovirsen Shows Phase III Success in Chronic Hepatitis B, GSK Confirms Bepirovirsen Shows Phase III Success in Chronic Hepatitis B, GSK Confirms Read Post »
Health TidingsNestlé Baby Formula Recall 2026: What We Know About the Recall & Cereu... Nestlé Baby Formula Recall 2026: What We Know About the Recall & Cereu... Read Post »
Clinical TrailsLilly’s Taltz Plus Zepbound Delivers Superior Outcomes in Phase 3b Psoria... Lilly’s Taltz Plus Zepbound Delivers Superior Outcomes in Phase 3b Psoria... Read Post »
Policy & AcquisitionsEli Lilly to Acquire Ventyx Biosciences for $1.2B to Expand Oral Inflammati... Eli Lilly to Acquire Ventyx Biosciences for $1.2B to Expand Oral Inflammati... Read Post »
Health TidingsVanda Therapeutics Receives Second FDA CRL for HETLIOZ in Jet Lag Disorder ... Vanda Therapeutics Receives Second FDA CRL for HETLIOZ in Jet Lag Disorder ... Read Post »
New Drug ApprovalFDA expands Cablivi approval to pediatric patients aged 12 and older with a... FDA expands Cablivi approval to pediatric patients aged 12 and older with a... Read Post »
New Drug ApprovalTakeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Takeda and Protagonist Submit NDA to FDA for Rusfertide in Polycythemia Ver... Read Post »
Policy & AcquisitionsFDA Narrows Oversight for Low-Risk Wellness Wearables, Clarifies What Stays... FDA Narrows Oversight for Low-Risk Wellness Wearables, Clarifies What Stays... Read Post »
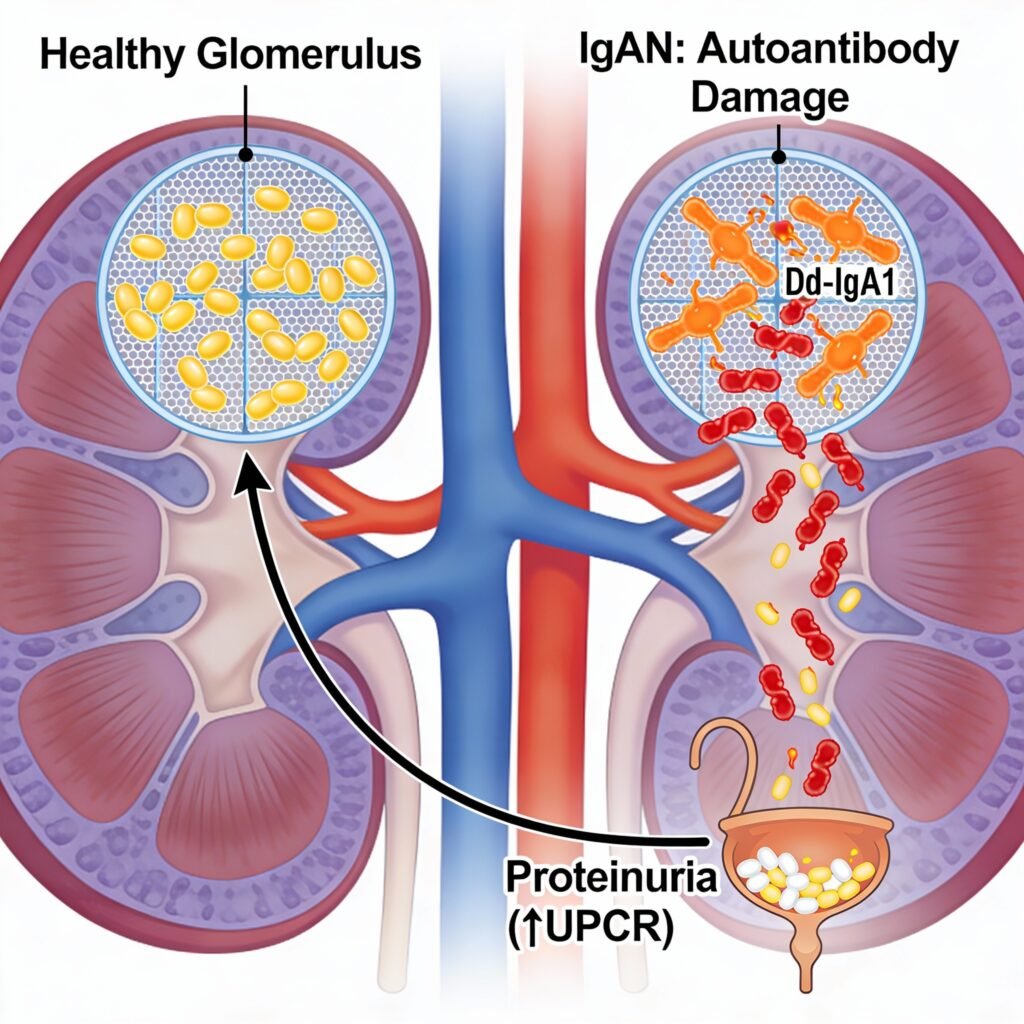
New Drug ApprovalFDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... FDA Grants Priority Review to Vera Therapeutics’ Atacicept for IgA Nephro... Read Post »
Clinical TrailsJohnson & Johnson Reports Positive Phase 2b JASMINE Trial Results for N... Johnson & Johnson Reports Positive Phase 2b JASMINE Trial Results for N... Read Post »
Policy & AcquisitionsMerck Completes Acquisition of Cidara Therapeutics to Advance Long-Acting P... Merck Completes Acquisition of Cidara Therapeutics to Advance Long-Acting P... Read Post »
Policy & AcquisitionsAmgen acquires Dark Blue Therapeutics to strengthen AML protein degrader pi... Amgen acquires Dark Blue Therapeutics to strengthen AML protein degrader pi... Read Post »
New Drug ApprovalJapan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Japan Approves Exdensur After U.S. and UK, Expanding Access for Severe Asth... Read Post »
New Drug ApprovalZai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Zai Lab’s AUGTYRO (repotrectinib) Receives NMPA Approval for NTRK-Positiv... Read Post »
New Drug ApprovalChina Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s China Accepts BLA for Subcutaneous LEQEMBI for Early Alzheimer’s Read Post »
Clinical TrailsFull Phase 3 TULIP-SC Results Support Self-Administered Saphnelo for Lupus Full Phase 3 TULIP-SC Results Support Self-Administered Saphnelo for Lupus Read Post »
New Drug ApprovalHealth Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Health Canada Approves REDEMPLO (plozasiran) for Familial Chylomicronemia S... Read Post »